Home
DOWNLOADs
USAGE
MOAT (Mutations Overburdening Annotations Tool) is a computational system for identifying significant mutation burdens in genomic elements with an empirical, nonparametric method. Taking a set of variant calls and a set of annotations, MOAT calculates which annotations have observed variant counts that are substantially elevated with respect to a distribution of expected variant counts determined by permutation of the input data. To produce this expected distribution, MOAT offers two types of permutation algorithm: one that permutes the locations of annotations (MOAT-a), and one that permutes the locations of variants (MOAT-v).
 Additionally, we provide a variant distribution simulator called MOAT-s, which produces permutations of the input variants, taking into account trinucleotide identity preservation, as well as the distribution of whole genome covariates
that influence the background mutation rate.
Additionally, we provide a variant distribution simulator called MOAT-s, which produces permutations of the input variants, taking into account trinucleotide identity preservation, as well as the distribution of whole genome covariates
that influence the background mutation rate.
Furthermore, MOAT offers integration with Funseq2 (funseq2.gersteinlab.org), a framework for evaluating the functional impact of single nucleotide variants. MOAT can compute the Funseq scores of
input variants alongside its permutation algorithms, enabling users to analyze both functional impact and mutation burden simultaneously. MOAT can also compute Funseq scores on the permuted variant datasets generated to simulate the
background mutation distribution, resulting in background Funseq scores to compare to the Funseq score of the observed input variants.
MOAT comes with parallel implementations for multi-CPU clusters and NvidiaTM CUDA-capable GeforceTM GPUs, enabling excellent runtime scaling with respect to the input size.
Here we provide download links for the MOAT source code and data context files necessary for MOAT analyses.
Download MOAT source code (zip archive, 612 KB)
Download Funseq2 whole genome signal track (compressed bigWig, 7.3 GB)
Download reference genome FASTA files (.tar.gz, 905 MB)
(Required for MOAT-v and MOAT-s)
Download mappability blacklist regions (.bed, 17 KB)
Download example annotation files:
Additionally, links for example inputs and outputs are available from the Example tab.
Table of Contents
Prerequisite SoftwareMOAT is developed for Linux-based operating systems. It is possible that MOAT may function under Mac OS X, or under Windows in an environment like Cygwin, but this has not been tested.
The following software are required to run MOAT. The "version tested" fields indicate the versions of each dependency that have been tested with MOAT. Earlier versions may work, but are unsupported.
1) gcc: The GNU project's C and C++ compiler. Necessary to produce executables from the C++ source code in the MOAT distribution.
Link: //gcc.gnu.org/
Versions tested: 4.4.7, 4.8.2
2) make: The GNU make utility, used to automate compilation of C++ source code in MOAT.
Link: https://www.gnu.org/software/make/
Version tested: 3.81
3) CUDA (For parallel MOAT-a only): Nvidia's Compute Unified Device Architecture framework for parallelizing computational workflows across graphics processing unit (GPU) stream processors. Requires an Nvidia GeForce or Quadro graphics card.
Link: //www.nvidia.com/object/cuda_home_new.html
Version tested: 6.5.12
4) OpenMPI (For parallel MOAT-v only): An open source Message Passing Interface implementation that facilitates interprocess communication between parallel processes.
Link: https://www.open-mpi.org/
Version tested: 1.10.1
Before MOAT can be used, its C++ source code must be compiled into executable binaries. All the requisite commands have been collected in the "makefile" file in the MOAT directory. To initiate C++ compilation, "cd" into the MOAT directory and run the "make" command.
Command summary:
cd [MOAT directory]
make
The makefile will compile all the serial source code, and detect any CUDA or OpenMPI installations, compiling the parallel source code if those frameworks are available. The makefile's output will indicate if the parallel versions are available for use.
1) hg19 reference genome
MOAT-v's algorithm depends on the human reference genome in order to preserve the trinucleotide context of the input variants when they are permuted. Therefore, MOAT-v requires a local copy of the human genome reference sequence FASTA files to operate. For our analyses, we used the hg19 FASTA files available from the UCSC Genome Browser at:
//hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
2) (OPTIONAL) Whole genome signal track file
MOAT-a and MOAT-v can retrieve precomputed whole genome signal scores for the input variants, which are combined into annotation scores based upon the sum of the scores of the intersecting variants. We provide precomputed Funseq scores on the MOAT website at:
//files.gersteinlab.org/public-docs/2017/04.21/funseq_hg19_wg.zip
These scores were generated by running Funseq2 over the entire human genome, saving the highest possible Funseq score at each location.
1) Overview
[] = user-defined parameter
{} = optional parameter
Usage: run_moat --algo=a --parallel=[y/n] -n=[number of permutations] --dmin=[minimum distance for random bins] --dmax=[maximum distance for random bins] --blacklist_file=[blacklist file] --vfile=[variant file] --afile=[annotation file] --out=[output file] --wg_signal_mode=[o/p/n] {--wg_signal_file=[whole genome signal file]}
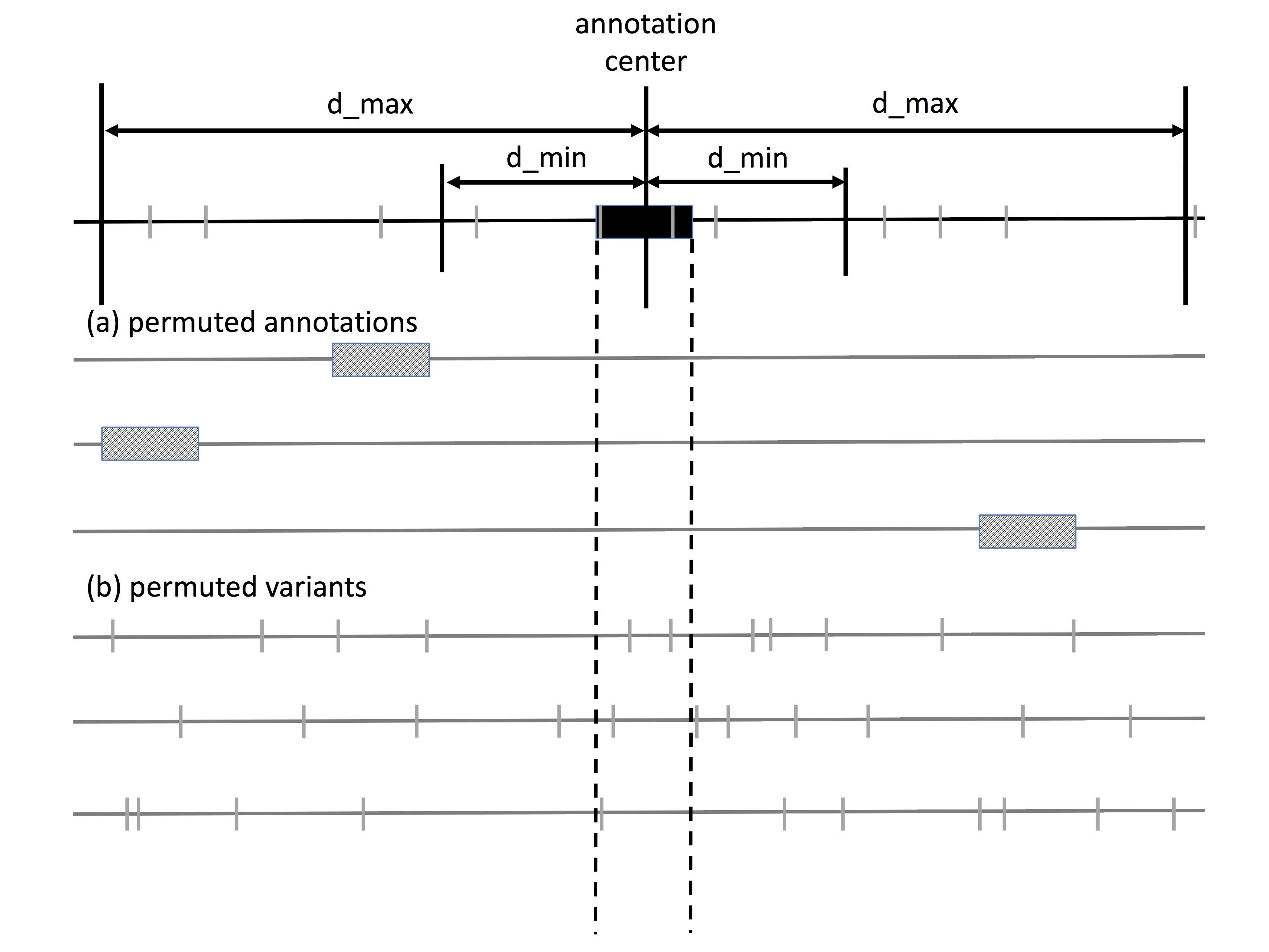
MOAT-a is the implementation of the annotation-based permutation algorithm. It iterates through the annotations in the [annotation file], and randomizes [number of permutations (n)] new locations for each annotation in the local genome context, whose boundaries are defined by [minimum distance for random bins (dmin)] and [maximum distance for random bins (dmax)]. Therefore, the local genome context is two intervals: one upstream of the annotation [-dmax, -dmin] and one downstream of the annotation [dmin, dmax]. Variants from the [variant file] are intersected with each of these permuted annotations to produce [n] permuted variant counts. The p-value of each annotation is the fraction of the permuted variant counts that are equal to or greater than the observed variant count (derived from intersecting the [variant file] variants with the annotation). These p-values are written to the [output file].
Additionally, [algo] specifies which permutation algorithm to use: [a] for MOAT-a and [v] for MOAT-v. The [parallel] flag indicates whether to use the CUDA-accelerated version [y] (recommended) or the much slower CPU version [n]. Additionally, the [blacklist file] is used to remove regions from consideration that have poor mappability, such as centromeres and telomeres, among others. Annotations that intersect the [blacklist file] will not be analyzed.
Furthermore, MOAT-a can provide the wg signal scores of the permuted variant datasets using the [whole genome signal file] provided. This option is enabled by setting [wg signal mode] to (p)ermuted variants mode. Signal scores computed from the permuted data will be compared to the scores of the observed data to derive the p-value significance of elevated scores. The second mode, (o)bserved variants only, computes the signal scores for the annotations as determined by the intersecting input variants, and outputs these scores alongside the mutation burden analysis results. The third mode, (n)o wg signal score analysis, will cause MOAT-a to only conduct a mutation burden analysis.
2) Input formats
Blacklist file
Tab-delimited columns: (chr, start, stop)
Extra columns are ignored.
Variant file
Tab-delimited columns: (chr, start, stop)
Extra columns are ignored.
Annotation file
Tab-delimited columns: (chr, start, stop, name)
Extra columns are ignored.
3) Output format
Output file
Tab-delimited columns: (chr, start, stop, name, p-value, wg signal score (optional), wg signal score p-value (optional))
1) Overview
[] = user-defined parameter
{} = optional parameter
Usage: run_moat --algo=v --parallel=[y/n] -n=[number of permutations] --width=[width of whole genome bins] --min_width=[minimum width of whole genome bins] --fasta=[reference genome FASTA file directory] --blacklist_file=[blacklist file] --vfile=[variant file] --out=[output directory] --ncpu=[number of parallel CPU cores to use] --3mer=[y/n] --wg_signal_mode=[y/n] {--wg_signal_file=[whole genome signal file]}
MOAT-v is the implementation of the variant-based permutation algorithm. It divides the human genome into bins of size [width of whole genome bins], and permutes variants within each bin. Essentially, [width] controls the size of the local genome context, in which we assume the covariates affecting the background mutation rate are more or less constant. The [min_width] parameter influences what MOAT-v will do when it produces bins less than [width], which can happen at the ends of chromosomes, or if a blacklist region is encountered as defined in the [blacklist file]. If the bin's size is higher than [min_width], MOAT-v will proceed with the bin as is. Otherwise, it will attempt to merge the bin with an adjoining neighbor bin to avoid having a bin size below [min_width]. If there are no adjoining neighbors available, the bin will be deleted.
Within each bin, variants from the [variant file] are shuffled to new locations to form [number of permutations (n)] permuted variant datasets. If [3mer] is set to [y]es, these new locations are chosen uniformly over the available trinucleotides that match the trinucleotide identity of the original variant. If [3mer] is set to [n]o, the new locations are chosen uniformly over all bin nucleotides. Trinucleotide identities are derived from the sequence data imported from the [reference genome FASTA file directory]. The [n] permuted variant datasets are produced as [n] files in the [output directory].
Additionally, MOAT-v can provide the wg signal scores of the permuted variant datasets using the [whole genome signal file] provided. This option is enabled by setting [wg signal mode] to (y)es. Otherwise, MOAT-v will only produce permutations for mutation burden analysis.
Furthermore, [algo] specifies which permutation algorithm to use: [a] for MOAT-a and [v] for MOAT-v. The [parallel] flag indicates whether to use the OpenMPI-accelerated version [y] (recommended) or the much slower single CPU version [n]. Finally, the [ncpu] option gives the user control over the number of CPU cores to use in the parallel version. This parameter can be omitted, in which case MOAT-v will automatically use all available CPU cores. This can also be achieved by setting --ncpu to MAX. Alternatively, the user can specify any number between 2 and the maximum number of cores available.
Using run_moat in this way will produce the permuted variant datasets, but to obtain annotation p-values, an additional program must be run: p_value_emp (explained further down)
NOTE: MOAT-v relies on the "bigWigAverageOverBed" program to retrieve precomputed wg signal scores. The 64-bit Linux version is provided with the MOAT distribution, but if you need another version, other versions are available from:
http://genome.ucsc.edu/goldenpath/help/bigWig.html (scroll to end of page)
Additionally, the use of the wg signal options will result in the generation of a temporary "tmp" directory in the MOAT directory. Therefore, if you attempt to run multiple instances of MOAT-v, this will likely cause the data interference between the instances, thereby corrupting the results. We recommend you clone the MOAT source code to another directory for parallel runs.
2) Input formats
Blacklist file
Tab-delimited columns: (chr, start, stop)
Extra columns are ignored.
Variant file
Tab-delimited columns: (chr, start, stop)
Extra columns are ignored.
3) Output format
Output directory
Files consist of tab-delimited columns: (chr, start, stop, wg signal score (optional))
4) Calculation of p-values
An additional program must be used after running MOAT-v to obtain p-values: p_value_emp
Usage: p_value_emp [variant file] [annotation file] [prohibited regions file] [permutation variants' directory] [output file] [wg signal option (o/p/n)] [wg signal file (optional)]
[variant file] is the same variant file used in run_moat, and the [annotation file] contains the annotations whose mutation burdens you want to evaluate. [prohibited regions file] works the same as [blacklist file], and [permutation variants' directory] corresponds to the [output directory] used in run_moat. The p-values are written to the [output file].
Additionally, p_value_emp can derive the Funseq scores of the annotations in the [annotation file], as implied by the variants in the [variant file]. [wg signal option] can be set to 'n' for (n)o Funseq, or 'o' for (o)bserved variants only, or 'p' for (p)ermuted variants. The observed variants mode computes wg signal scores for the [variant file] variants, sums them over the annotations in the [annotation file], and adds them to the [output file]. The permuted variants mode does the same thing as observed variants mode, and also uses the wg signal scores for the permuted variant datasets (must have used wg signal score computation in MOAT-v) to calculate a p-value significance for the annotations' wg signal score, which are calculated and reported similar to the mutation burden p-value. All score information is derived from the [wg signal file].
p_value_emp's [output file] has the following tab-delimited columns:
(chr, start, stop, name, p-value, wg signal score (optional), wg signal score p-value (optional))
1) Overview
[] = user-defined parameter
Usage: run_moat --algo=s --parallel=[y/n] -n=[number of permutations] --width=[width of whole genome bins] --min_width=[minimum width of whole genome bins] --fasta=[reference genome FASTA file directory] --blacklist_file=[blacklist file] --vfile=[variant file] --out=[output directory] --ncpu=[number of parallel CPU cores to use] --3mer=[y/n] --covar_file=[covariate signal file 1] [--covar_file=[covariate_signal_file_2] ...]
MOAT-s is a somatic variant simulator that produces [n] permutations of the input [variant file]. Like MOAT-v, it divides the human genome into bins of size [width of whole genome bins]. [width] controls the size of the local genome context, in which we assume the covariates affecting the background mutation rate are more or less constant. The [min_width] parameter influences what MOAT-s will do when it produces bins less than [width], which can happen at the ends of chromosomes, or if a blacklist region is encountered as defined in the [blacklist file]. If the bin's size is higher than [min_width], MOAT-s will proceed with the bin as is. Otherwise, it will attempt to merge the bin with an adjoining neighbor bin to avoid having a bin size below [min_width]. If there are no adjoining neighbors available, the bin will be deleted.
Bins are clustered using k-means clustering based on the similarity of their covariate signal profiles derived from the [covariate signal files]. At least one such file must be provided when invoking MOAT-s. If [3mer] is set to [y]es, the variants from the [variant file] are relocated within their cluster's bins to locations with the same trinucleotide identity as their original sites. If [3mer] is set to [n]o, new locations are chosen uniformly over all nucleotides in the bin cluster. Trinucleotide identities are derived from the sequence data imported from the [reference genome FASTA file directory]. The [n] permuted variant datasets are produced as [n] files in the [output directory].
The [parallel] flag indicates whether to use the OpenMPI-accelerated version [y] (recommended) or the much slower single CPU version [n]. Finally, the [ncpu] option gives the user control over the number of CPU cores to use in the parallel version. This parameter can be omitted, in which case MOAT-s will automatically use all available CPU cores. This can also be achieved by setting --ncpu to MAX. Alternatively, the user can specify any number between 2 and the maximum number of cores available.
NOTE: MOAT-s relies on the "bigWigAverageOverBed" program to generate the covariate signal profiles for the whole genome bins. The 64-bit Linux version is provided with the MOAT distribution, but if you need another version, other versions are available from:
http://genome.ucsc.edu/goldenpath/help/bigWig.html (scroll to end of page)
Additionally, the use of "bigWigAverageOverBed" will result in the generation of temporary files in the MOAT directory. Therefore, if you attempt to run multiple instances of MOAT-s, this will likely cause the data interference between the instances, thereby corrupting the results. We recommend you clone the MOAT source code to another directory for parallel runs.
2) Input formats
Blacklist file
Tab-delimited columns: (chr, start, stop)
Extra columns are ignored.
Variant file
Tab-delimited columns: (chr, start, stop, ref, alt)
Extra columns are ignored.
3) Output format
Output directory
Files consist of tab-delimited columns: (chr, start, stop, ref, alt)
The data available here contains a single variant file and a single annotation file, which may be used to verify that MOAT is running correctly. The variant file contains some ~8 million randomly generated single nucleotide variants, and the annotation file consists of a set of transcription start sites (TSS) parsed from a GENCODE file.
To study the mutation burdens of these TSSes with MOAT-a, these two files are given to the run_moat program as the vfile and afile, respectively. We provide a blacklist file in the Downloads tab, although the user is free to define his or her own. Aside from the file parameters, the choice of the number of permutations n, the minimum range for sampling random bins d_min, and the maximum range for sampling random bins d_max, are important for producing meaningful MOAT-a output. Precise p-value calculation depends on running a large number of permutations, and we typically use n=10,000. The choice of d_min and d_max depend on the size of the annotations in the afile. For TSSes of ~100bp size, we set d_min=2000 so that it is substantially large enough to avoid signal bleed from the target annotation. d_max is typically chosen to be large enough to allow sufficient sampling range, but also small enough that the mutation covariates are roughly constant throughout the sampling region. For TSS analyses, we use d_max=100,000.
Sample MOAT-a call:
./run_moat --algo=a --parallel=y -n=10000 --dmin=2000 --dmax=100000 --blacklist_file=blacklist.combined.3col.sort.merge.bed --vfile=example_variants.txt --afile=tss.bed --out=sample_out.txt --wg_signal_mode=n
In MOAT-v and MOAT-s, the technical runtime parameter choices include the number of permutations n, the width of the whole genome bins width, and the minimum bin width min_width. The bin width is selected to be small enough that the mutation covariates are roughly constant throughout the region, enabling the assumption of a constant mutation rate to hold. min_width is about ensuring that, should a bin be truncated due to overlap with the blacklist regions, it will not drop below a certain width to guarantee sufficient choices for permuted variant placement. min_width is typically set to be half the width. Additional file parameters include the whole genome FASTA reference files, and any covariate files to be used with MOAT-s. Also, if run in parallel on multiple CPU cores, specify the number of CPU cores to use with ncpu. Finally, specify the use of the trinucleotide context preservation feature with 3mer.
Sample MOAT-v call:
./run_moat --algo=v --parallel=y -n=10000 --width=100000 --min_width=50000 --blacklist_file=blacklist.combined.3col.sort.merge.bed --fasta=hg19_fasta --vfile=example_variants.txt --out=tss.bed --3mer=y --wg_signal_mode=n --ncpu=16
Sample MOAT-s call:
./run_moat --algo=s --parallel=y -n=10000 --width=100000 --min_width=50000 --blacklist_file=blacklist.combined.3col.sort.merge.bed --fasta=hg19_fasta --vfile=example_variants.txt --out=tss.bed --3mer=y --wg_signal_mode=n --ncpu=16 --covar_file=replication_timing.bw
Here we provide download links for a fully worked out MOAT example run, which was used to evaluate MOAT cancer driver detection capability.
Variant file
We used cancer variant data published by Alexandrov et al. (2013) available here. For our evaluation, we used the Breast cancer subset.
Annotation file
We used MOAT to evaluate somatic mutation burdens in a set of transcription start sites (TSSes) derived from taking the 100bp regions immediately upstream of coding transcripts, according to GENCODE. This set of annotations is available here.
Blacklist file
We used the same blacklist file featured on the Downloads page, available here.
MOAT command line
./run_moat --algo=a --parallel=y -n=10000 --dmin=2000 --dmax=100000 --blacklist_file=blacklist.combined.3col.sort.merge.bed --vfile=breast.snv.bed --afile=coding.tss.bed --out=out.txt --wg_signal_mode=n
Output
The output of this command is available here.